
文末可领取免费 PCR 进阶教学资源
PCR 是一个坑,埋了好多研究僧……
PCR 实验中常遇见一些令人头疼的小问题,可能就是实验设计或者实验过程中一个小小的条件没有到位,就导致预期的实验结果出不来。
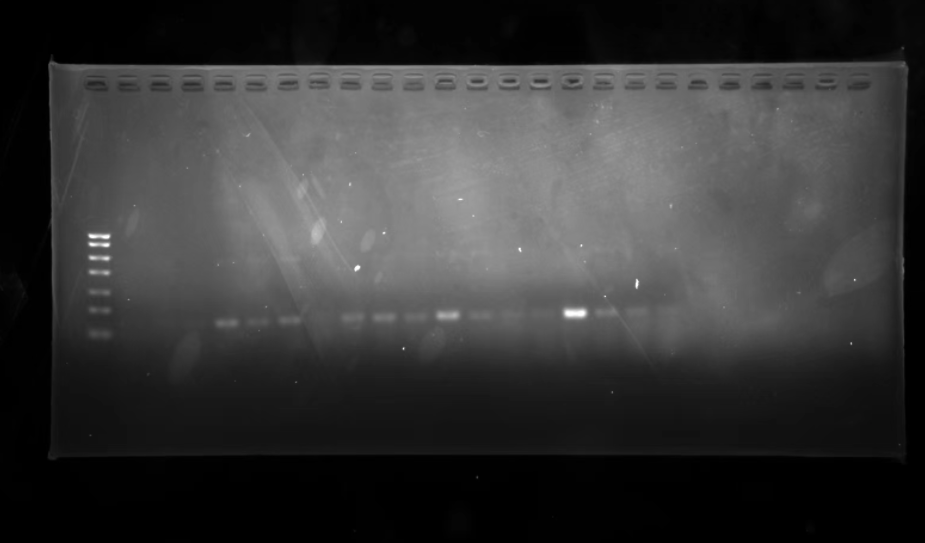
PCR 总是 P 不出,往往有 3 个原因,来看看究竟是哪一环节出了错吧。以下内容来自丁香实验「PCR 专题」,超全 protocol 经验总结图文不断更新,建议点击收藏!
戳此收藏订阅 PCR 专题👇👇👇
 样本制备你的 RNA 样本合格吗?
样本制备你的 RNA 样本合格吗?常见的 RNA 提取方法有 TRIzol 法、吸附法,这两个方法各有所长,但都不能保证提取的 RNA 样本一定合格。
因而,提取完成后应测定 RNA 的浓度及吸光度。通常,在 260,280,230 nm 处分别测定其吸光度(A260、A280、A230)并计算其比值(A260 /A280,A260 /A230)。

230/260/280 究竟有何意义?
A260 为核酸的吸光度,A280 为蛋白质的吸光度,A230 为其他杂质(多糖等)的吸光度。纯 DNA 的 A260 /A280 为 1.8,纯 RNA 的 A260 /A280 为 2.0。
如果 OD 比值不在范围内该如何解决呢?
再次洗涤样本!
75% 乙醇沉淀 RNA 后重新吸附、洗涤;若采用 TRIzol 法提取 RNA,可直接洗涤两次。洗涤后离心时可两次分别将 EP 管置于不同的方向,便于彻底洗涤 RNA 样本。通常洗涤两次后可使测得的 OD 值较为理想。
引物特异性太差试试这个免费软件提到 PCR 老是 P 不出,大部分同学都会觉得是引物的锅。
引物,是进行 PCR 实验的必备条件。其特异性的问题可通过「BLAST」搞定,让人比较头痛的是引物可以形成「发夹结构」或「二聚体」(即在<75℃ 时出现溶解曲线的峰),影响扩增效率,最终导致实验结果不可用。
那么,这个问题能否解决呢?
有!设计引物的时候用「RNAstructure」预测一下引物的二级结构即可。

那么,这个软件要如何使用呢?文章篇幅有限,大家可以点击下方卡片查看全部教学内容👉 点此查看软件教学
最后一步保证扩增效率好多同学反馈:同一批样本、同一批引物、甚至都用一起配的胶了,P 出来的结果依旧不!一!样!真的很崩溃,但这其实不是操作者的问题。
不知道各位看官有没有遇到类似的问题?先说说出现这类问题的原因吧:
1. 扩增条件非最佳;
2. 样本中含有抑制扩增的物质,如乙醇。
一对引物有其特定的 Tm 值,但 Tm 值时未必是其最佳扩增温度。温度过高时,扩增效率过低,差异部分的目的基因不能被扩增,导致「假阴性」结果的出现。那么,如何确定扩增效率是否满足要求呢?
通常,拿到新引物后应从 Tm- 5℃ 寻找其适宜的扩增温度,在某温度起为特异性扩增。
然后,在特异性扩增的温度下检测扩增效率:将 cDNA 梯度稀释(一般为 2×),测试稀释后的样本 Ct 值差异是否为 1 左右。
1、若差值为 1,则扩增效率良好,可进行正式实验。
2、若在特异性扩增的温度范围内不能使 Ct 值差异为 1,则需考虑重新设计引物。
以上内容来自丁香实验「PCR 专题」,超全 protocol 经验总结图文不断更新,建议点击收藏!
如果你有更多实验问题欢迎进入【丁香实验小程序】查看、提问!
戳此收藏订阅 PCR 专题👇👇👇
一起看看 PCR专题包含哪些内容01-保姆级教学视频
02-超多 Protocol

03-经典实验问答
04-延伸图文阅读
丁香实验做更优质的实验内容资讯离不开各位科研 er 的支持和关注!如果你是师兄师姐,也欢迎把本篇文章分享给师弟师妹们!
扫码加入【PCR学术交流群】
PCR专题学习资料免费学
科研试剂/试剂盒/科研器材免费试
👇👇👇
策划|Olaf封面|小黄做的 PCR配图|丁香实验设计团队